【概要】
次世代シーケンサと呼ばれる技術の進歩により、ゲノムのDNAや、DNAを転写したRNAなどの塩基配列の情報を従来にくらべて飛躍的に 低いコスト、短時間で得ることができるようになりつつある。なかでも米国イルミナ社のゲノムアナライザは情報量に対するコストパーフォーマンスが高く、現 時点でもっとも広く普及している機種となっている。奈良先端科学技術大学院大学(学長:磯貝彰)情報科学研究科計算システムズ生物学研究室の中村建介特任 准教授と金谷重彦教授らは、現在のイルミナ社の解析データには読み取ることの難しい配列パターンが存在することを世界に先駆けて見いだした。この情報に基 づいて、イルミナシーケンサの高い能力を最大限に引き出すことで、SNPs変異(1つの塩基だけ異なる変異)の特定や、未知ゲノムの再構築を、これまでよ り高い精度で行うことが出来るようになる、と期待される。
【解説】
ゲノムDNAなどの塩基配列の解読技術が近年急速に進んでい る。ほんの十年ほど前にはヒトゲノムプロジェクトに代表されるように国家プロジェクト規模の予算と人材(数年の時間・数十人・数十億円)を必要とした情報 が、次世代シーケンサと呼ばれる装置を用いることにより研究室規模(数日・数名・数十万円)で得られるようになっている。その結果、たとえば個人のDNA 配列のどの部分が標準的なヒトの塩基配列と異なっているかを簡便に調べることが出来る。こうした情報と、疾患や薬に対する副作用の感受性などの情報を組み 合わせれば、効果が高く副作用の低い「テーラーメード医療」とよばれるような個々人の体質に合わせたきめ細かい医療の実現に大きく貢献できる。
ま た、数々のゲノムプロジェクトにより既にいくつかのモデル生物種についてゲノム配列が決定されているが、これらのゲノム配列情報の活用を進めてゆくポスト ゲノム研究においては、モデル生物にある程度類似していて、形質・機能がわずかに異なる近縁生物種のゲノム配列を特定することが、進化の研究などに有益な 情報を与えてゆくと考えられる。このような基礎生物学におけるゲノム情報の機能解析においても次世代シーケンサから得られるデータは重要な役割を果たす。
次 世代シーケンサと呼ばれるテクノロジーの中でイルミナ社のシーケンサは現在もっとも普及している機種であるが、得られる配列情報についていくつかの問題点 が存在する。たとえば、イルミナ社自身の見解として、イルミナシーケンサにより特定された未知のSNPs(一塩基変異)については、他の実験手法を用いて 確認することが必要であるとアナウンスされている。結果、配列解析の結果として新しい変異を見いだしたとしても、他の実験手法による検証が必要とされるた め価値が半減してしまう。
私たちは、このような検証が必要とされる理由がどこにあるのかを疑問に思いつつシーケンシングデータの解析を進めてゆく上で、イルミナシーケンサの配列データに共通する興味深い特徴が存在することを見いだした。
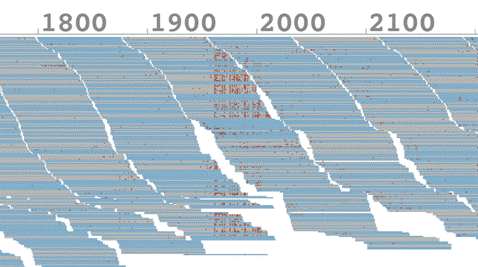
イ ルミナ社のシーケンサによる配列データの精度は実験の条件等にもよるが1%程度と言われている。既知のバクテリアゲノムに対してシーケンサにより得られた データをマップした結果の一部分を図1に示す。それぞれの図の上部の数値は既知ゲノム配列上の塩基位置を表しており、その下に積み重なった短い線分がシー ケンサから得られたリードと呼ばれる一つ一つの配列データ(各75塩基長、DNAの断片)を適合するゲノム位置にマップしたものである。配列データの色は シーケンシング時の読み取り方向とゲノム方向が一致している場合には薄い灰色、逆向きの相補鎖として一致する場合には薄い青色で示されている。
また、それぞれのリードは標準データのレファレンスゲノムに対して最大35箇所のミスマッチまで許容して、レファレンス上で最もミスマッチがすくなくなる位置にマップされている。
従 来のマッピングプログラムを用いた場合には、許されるミスマッチの数はリードあたり2個程度でギャップアラインメント(ギャップ調整)と呼ばれる短い塩基 の挿入と欠損を考慮した手続きがとられるが、今回、私たちは多くのミスマッチを許しながら、ギャップアラインメントを行わないマッピングを行うことで、図 1に見られるように、レファレンスとのミスマッチが集中して発生する領域が存在することを見いだした。
さらにこの図で興味深いのは、赤い 点で示されたミスマッチが右方向へ読み取っている灰色のリードに集中していて、逆方向の薄い青色のリードにはあまり見られないこと。またミスマッチが 1960塩基位置付近を境界として右側、すなわち読み取り方向に特定の開始点が存在しているように見えることである。このことから配列読み取りにエラーを 引きおこす要因が1950塩基位置付近に存在することが示唆される。私たちはこの配列特異的な読み取りエラーをSSE(Sequence Specific Error)と名付け、その発生のメカニズムの推定とこれにより引きおこされるシーケンシング上の問題点を整理して今回の論文で報告した。
図 1. 配列特異的エラー(SSE)の発生位置、上部の数値はゲノム位置。赤い点はゲノム塩基とマップされたリードの塩基が異なる箇所を示す。1960塩基付近を 境界として、右側(読み取り方向)に特定の開始点が存在しているとみられ、エラーの要因が1950塩基位置付近に存在すると示唆された。
図2. 左上:サンプルとレファレンスの間に一塩基置換(SNP)が起きている例 左下:SSEによりSNP
のようにミスマッチが起きている例 右 :複数のSSEにより読み取りが困難になっている領域
今 回特定されたエラーパターンによる主な問題として(1)一塩基置換(SNPs)の誤認、(2)RNA-seqという方法による発現量を調べる解析などで、 リード数から発現量を見積もる場合の定量性、(3)ゲノム配列の再構築(アセンブル)において読み取り困難な領域が存在することによる不連続領域―が挙げ られる。
(1)については図2に示したように、真の一塩基置換が左上の図に示したように現れるのに対して、SSEによる読み取りエラーが 左下図の様に約半数のリード配列で同じ塩基部位に現れることがある。このようなエラーはたとえば真核生物の読み取りにおいてはハプロタイプ(片親由来の DNA塩基配列)の一方に一塩基置換が存在すると誤認される可能性が高い。図2右側ではSSEによるエラーが狭い箇所に集中して起きているため、ミスマッ チを多く許したマッピングでは上のように多くのミスマッチが現れ、従来のプログラムで通常のマッピング条件によりミスマッチを2つしか許さない場合には リードがほとんど張り付かない状態(図2右下)を示している。
(2)では、細胞中に発現しているRNA配列の相対量をRNAを鋳型として 作成したDNA配列のフラグメントの存在量から推定する手法において、図2の右側のようにSSEに起因するエラーが多く含まれる配列領域を含むトランスク リプト(転写物)が存在する場合には、その発現量を過小評価してしまう可能性がある。また、このような領域の存在は(3)のゲノム配列の再構築をしようと する場合、イルミナシーケンサーデータからは配列を再現することが事実上不可能な不連続領域となってしまう。
【本研究の意義】
私 たちの特定したイルミナシーケンサのエラープロファイルは、図2で示したように配列解析においてさまざまな問題の要因となっている。このエラープロファイ ルについて、基本的なメカニズムの推定ができていることから、今後、原因配列の特定を進めることで読み取りエラーが起きやすい領域を予測し、さらにはエ ラー発生のモデルを考慮して塩基決定(ベースコール)のアルゴリズムを設計することで、高性能なイルミナ社のシーケンサのデータを最大限に引き出すことが 出来るようになる、と期待される。
【補足説明】
1. 次世代シーケンサー:従来のSanger法と呼ばれる塩基配列解析技術に対して、短い塩基配列を大量に読み出すことの出来る配列解析プラットフォームの総 称。代表的なものとして(1) Illumina Genome Analyzer, (2) Life Technologies/ABI SOLiD System, (3) Roche/454 Genome Sequencer FLXの3つが広く使われている。
2. SNPs:一塩基多型、ある生物集団に一定の割合で存在する一塩基置換
3. マッピング:シーケンサから得られた短い塩基配列データ(リード)を既知のレファレンス塩基配列上で最も矛盾無く対応すると思われる場所にあてはめてゆく操作。アラインメントとも呼ばれる
4. アセンブル:シーケンサから得られた短い塩基配列データを、重複配列をのりしろとして重ね合わせながら伸張し、元の塩基配列を再構築しようとする操作。